Downloading and organizing files
Last updated on 2024-09-24 | Edit this page
Overview
Questions
- How do we process raw RNAseq reads to generate count data?
- What are the key steps involved in quality control, alignment, and quantification of RNAseq data?
- How do different tools and parameters affect the outcome of RNAseq data processing?
- Why is it important to ensure accurate and consistent read mapping and quantification?
Objectives
- Participants will learn the essential steps from raw read quality control to the generation of count data.
- Hands-on practice with
FastQCto assess the quality of raw reads. - Learn how to use
STARto map reads to a reference genome. - Practice using
featureCountsfromsubreadspackage to generate count data from aligned reads. - Develop the ability to assess the quality and consistency of the resulting count data, identifying and addressing common issues.
Introduction
In RNAseq analysis, transforming raw sequencing data into meaningful insights requires a series of well-defined steps. This episode will guide you through the essential process of converting raw RNAseq reads into count data, which forms the basis for downstream analyses such as differential expression and functional annotation.
We will start with quality control to ensure that the raw data is suitable for analysis, followed by the alignment of reads to a reference genome or transcriptome. Finally, you’ll learn how to quantify gene expression levels by counting the aligned reads associated with each gene. Through hands-on exercises, you’ll gain practical experience with the tools and techniques that are critical for accurate RNAseq data processing.
Firstly, the reads in FASTQ format are usually stored or transferred
in compressed formats, often with an extension of .fastq.gz
or .fq.gz. This compression helps save storage space and
reduces the time required for data transfer.
Should I Uncompress fastq files?
Uncompressing FASTQ files is often unnecessary since many RNAseq
tools can directly process compressed files, such as those in
.gz format. Keeping FASTQ files compressed not only saves
storage space but also reduces the time and computational resources
required for data handling. It’s generally more efficient to work with
compressed files, especially when dealing with large datasets.
Secondly, library preparation steps of RNAseq usually add adapters and other technical sequences that may be present at the ends of reads.
Should I trim adapters for RNAseq analyses?
For most RNAseq analyses, trimming adapters is not necessary, as many popular mapping programs, like HISAT2 and STAR, can effectively soft-clip adapter sequences during the alignment process. However, if you are performing more specialized analyses, such as transcript assembly or other applications where read length consistency is crucial, trimming adapters may be important. Aggressive trimming might actually negatively impact expression analyses! More information here.
For this lesson, we will use a dataset from the experiment
Downloading the data and organization
We will begin by downloading the raw reads for our experiment from NCBI GEO database. In real world, you will probably obtain your raw reads from the sequencing center or from your collaborators, but for this lesson, we will download the pre-analyzed data from a public repository.
What files do I need for RNAseq analysis?
The essential files for RNAseq analysis include raw reads, reference genome, and annotation files. Raw reads are typically provided in FASTQ format, which contains the nucleotide sequences and quality scores of the reads. The reference genome is typically provided in FASTA format, which contains the nucleotide sequences of the chromosomes or contigs. The annotation file is usually in GTF (Gene Transfer Format) or GFF (General Feature Format) format, which provides information about the genomic features such as genes, transcripts, and exons.
Maintaining this organized structure ensures a streamlined workflow, reduces the risk of errors, and facilitates easier access to files throughout the analysis process.
Create the directories needed for this episode
Create a directory to serve as the working directory for the rest of
this episode and lesson (the workshop example uses a directory called
rcac_rnaseq). Then, within this chosen directory, create
the four fundamental directories previously discussed
(data, scripts, mapping, and
counts).

Downloading the data
The data files include the raw reads, reference genome, and
annotation files. We will download them to the data
directory in our working directory.
Where will I find the data required for my project?
The raw reads for your RNAseq experiment will typically be provided by the sequencing facility or your collaborators. The reference genome and annotation files can be downloaded from public repositories such as Ensembl, UCSC Genome Browser, or Gencode or NCBI. There are also specialized databases for several organisms, such as FlyBase, WormBase, TAIR, and MaizeGDB for other model organisms.
The raw reads were generated from a mouse model, and we will use the
mouse reference genome and annotation files for mapping and
quantification. We will use GenCode’s GRCm39
reference genome and annotation files. Once we have the link, we can use
wget to download the files.
Annotation
Take a look at the GenCode’s GRCm39 website. There are few choices for annotation files (and formats). What annotation file (and format) will you download?

Reference genome
Similarly, what reference genome file will you download?
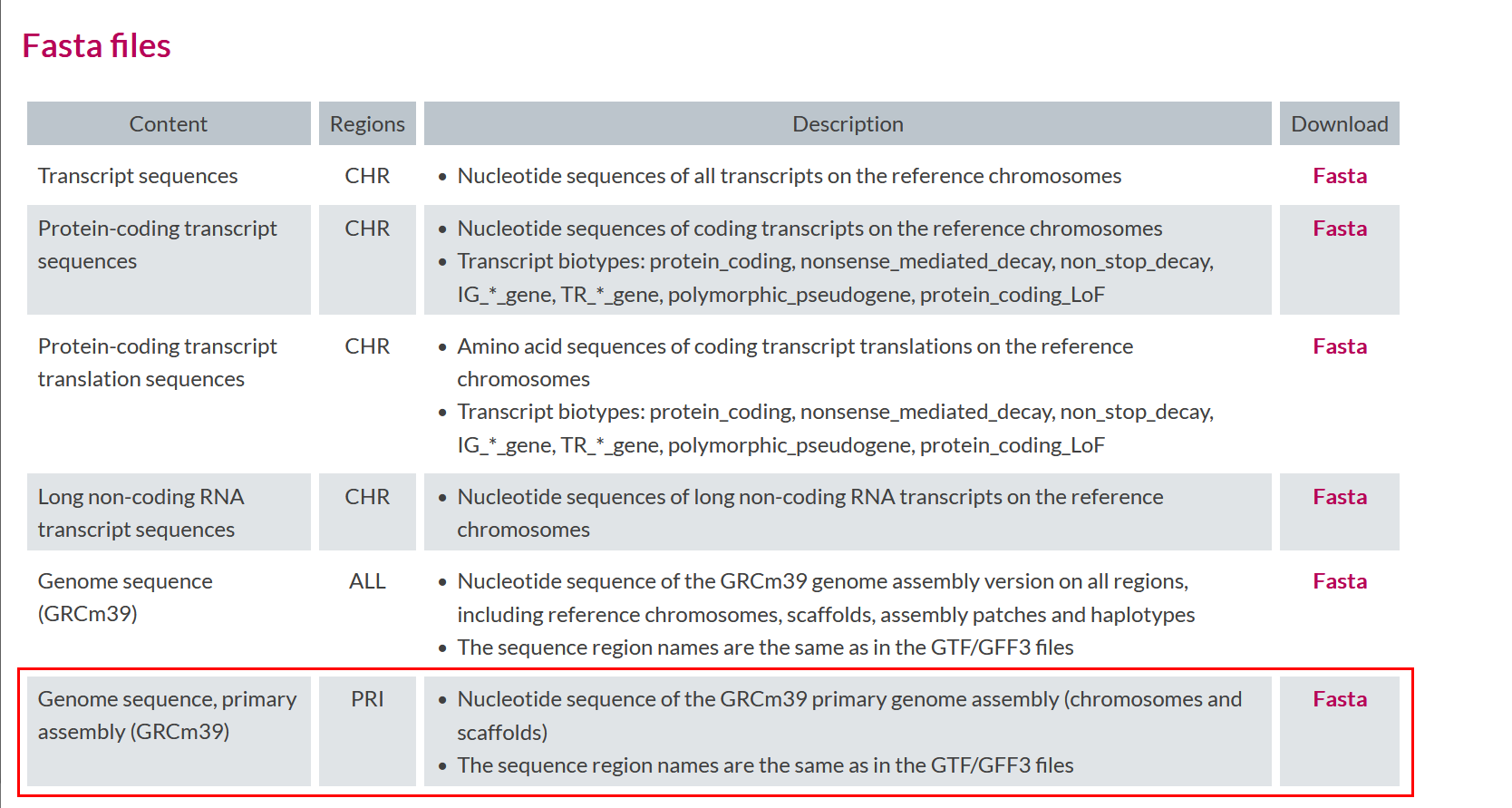
Commands to donwload the files are as follows:
BASH
cd rcac_rnaseq
GTFlink="https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M35/gencode.vM35.primary_assembly.basic.annotation.gtf.gz"
GENOMElink="https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M35/GRCm39.primary_assembly.genome.fa.gz"
# Download the reference genome
wget -P data ${GENOMElink}
# Download the annotation file
wget -P data ${GTFlink}
# Check the files
cd data
ls
# Uncompress the files
gunzip GRCm39.primary_assembly.genome.fa.gz
gunzip gencode.vM35.primary_assembly.basic.annotation.gtf.gzRaw reads
The raw reads for RNA-seq data required for the subsequent episodes were generated to investigate the impact upper-respiratory infection have on changes in RNA transcription in the cerebellum and spinal cord of mice. This dataset was produced as part of the following study:
Blackmore, Stephen, et al. “Influenza infection triggers disease in a genetic model of experimental autoimmune encephalomyelitis.” Proceedings of the National Academy of Sciences 114.30 (2017): E6107-E6116.
The dataset is available at Gene Expression Omnibus (GEO), under the accession number GSE96870.

Where do you find FASTQ files?
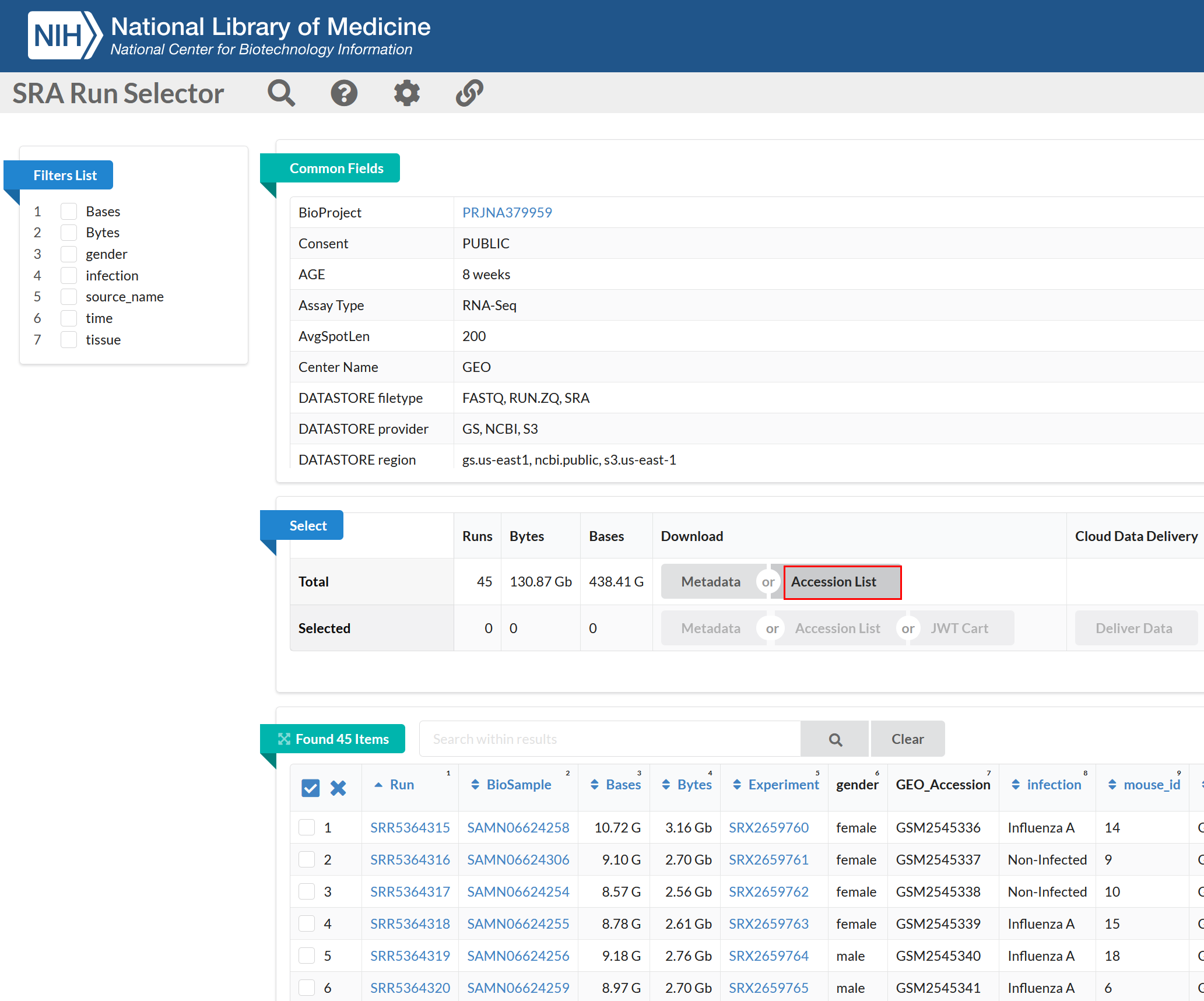
GEO provides information about the experiment and the raw data files accessions. This information is needed for understanding the experimental design and contrasts you need to make to get useful biological insights. Although GEO provides the metadata, the dataset is actually hosted in the SRA (Sequence Read Archive) database. How do you find the raw reads for this experiment?
Follow the SRP102241
link and then click on the Send results to Run selector to
get the list of runs. This page will provide you with the list of runs
associated with the experiment. Clicking on the “Accession List” will
download all the SRR ids you need.
For downloading the raw reads, we will use the
fasterq-dump tool from the SRA Toolkit. The following
command will download the raw reads for the experiment:
BASH
module load sratoolkit
while read SRR; do
fasterq-dump -O data ${SRR}
done < data/SRR_Acc_List.txtAs there are many files to download, and the process may take some
time, we will use the previously downloaded files for the rest of the
episode. The files are located in the depot directory and
can be soft-linked to the data directory.
Key Points
- Raw reads are typically provided in FASTQ format, which contains the nucleotide sequences and quality scores of the reads.
- Reference genome and annotation files are essential for mapping and quantification of RNAseq data.
- Organizing data files in a structured manner facilitates efficient data handling and analysis.
- Tools such as
wgetandfasterq-dumpcan be used to download data files from public repositories.